Scott Prescott, MD/PhD
Professor
Department of Physiology
University of Toronto
Senior Scientist, Neurosciences & Mental Health
Hospital for Sick Children
(Dec. 7, 2022)
Somatosensory Coding Gone Wrong — the Origins of Neuropathic Pain
Who hasn't been told after stubbing a toe or bumping an elbow, "rub it and it will feel better?" But what if that had the opposite reaction? What if the pain never went away? Such is the experience of neuropathic pain: pain without direct cause and without easy remedy. Dr. Prescott discussed his work looking for the sources of this unexplained pain. His lab has identified that changes in ion channels, tunnels in the cell membrane that allow for substances such as sodium, potassium and calcium to enter or exit, can lead to hyperexcitability, a cell that can’t turn off. This, Dr. Prescott suggests, is one part of a system responsible for chronic pain.
Recall a bad sunburn. Imagine the searing pain caused by your shirt collar touching your neck or the shower water on your back. Stimuli that normally attract no attention, or that should be soothing, now make you recoil in pain. If the sunburn was instead a nerve injury, the same hypersensitivity and spontaneous burning pain might develop, but never relent. Chronic pain caused by damage to the nervous system (neuropathic pain) affects one in 10 people and is notoriously difficult to treat. Unlike "normal" pain, neuropathic pain exists in the absence of noxious sensory input, which suggests that neural processing within the somatosensory system has somehow gone awry. But how? And how do we fix it?
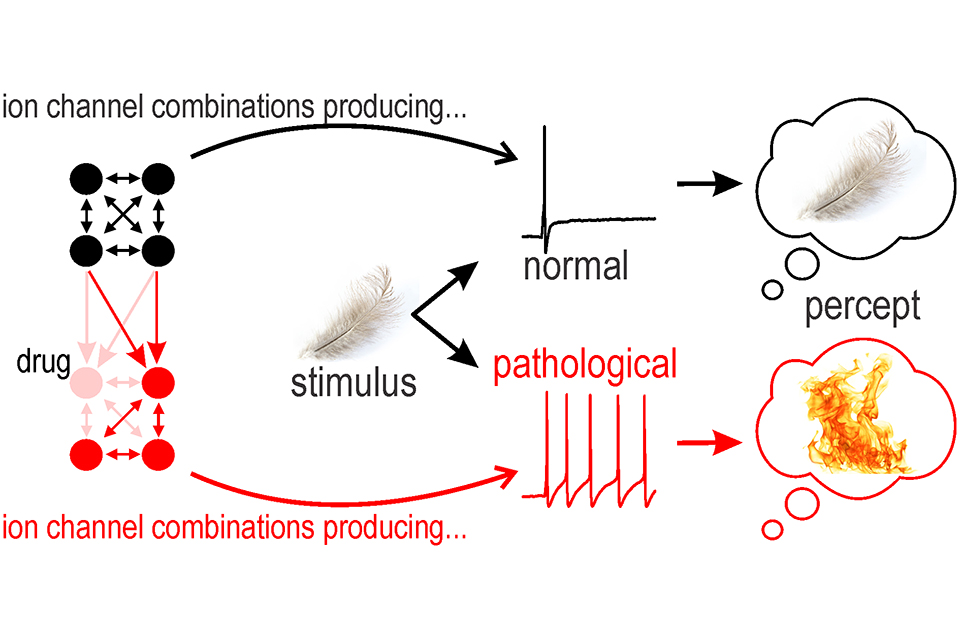
Nearly all neurons rely on action potentials (spikes) to encode, process, and transmit information. Excitability — the capacity to generate spikes — depends on the interaction between diverse ion channels. In neuropathic pain, some sensory neurons become hyperexcitable, spiking too much and in abnormal patterns. To explore this, we experimentally reproduced injury-induced ion channel changes by blocking certain channels pharmacologically or adding other channels virtually with dynamic clamp. We found that that equivalent hyperexcitability can arise through distinct ion channel changes. This degeneracy has important implications for how neuropathic pain develops and how to treat it; for instance, we found that chronic blockade of Nav1.7 channels leads to upregulation of other sodium channels, thereby subverting effects of Nav1.7-specific antagonists.
Distorted by changes in sensory neuron excitability, somatosensory information is relayed from the periphery to the spinal cord, where other injury-induced changes have taken place. In particular, synaptic inhibition is compromised by changes in intracellular chloride regulation. Whereas touch signals normally inhibit pain signals, disinhibition corrupts this interaction, allowing touch to cause pain instead. Computer modeling of the spinal circuit can reproduce those changes. Exploring that model reveals that equivalent circuit function can be achieved with different sets of synaptic weights — another example of degeneracy. This raises concerns that therapeutically targeting certain spinal cell types might face the same challenges as targeting specific ion channels — degenerate systems can compensate. Therapeutically targeting one part of a complex system without regard for how the rest of the system will compensate is liable to produce disappointing results. A more integrative approach promises to yield better treatments for chronic neurological disorders, like neuropathic pain.